หลักเกณฑ์การจัดทำคุณลักษณะเฉพาะของยา
1. บทนำ
ตามที่กระทรวงสาธารณสุขได้มีนโยบายที่ชัดเจนในการพัฒนาประสิทธิภาพระบบบริหารเวชภัณฑ์โดยได้ มีการปรับปรุงแนวทางการปฏิบัติเพื่อให้เกิดความชัดเจน โปร่งใส ตรวจสอบได้ และให้มีการพัฒนาอย่างต่อเนื่องและสอดคล้องกับสถานการณ์ ทั้งทางด้านการบริหารงาน การจัดสรรงบประมาณในการจัดซื้อ ระบบบริหารเวชภัณฑ์ รวมถึงระบบการตรวจสอบและระบบรายงาน
มาตรการหนึ่งของการเพิ่มประสิทธิภาพในการบริหารเวชภัณฑ์ดังกล่าวเพื่อให้ได้มาซึ่งเวชภัณฑ์ที่ดี มีคุณภาพและลดต้นทุนในด้านการบริหารจัดการ สำหรับคุณภาพของเวชภัณฑ์ที่จะจัดหานั้น กล่าวได้ว่า เป็นหัวใจของการจัดหาเวชภัณฑ์ เนื่องจากการได้เวชภัณฑ์ที่ไม่มีคุณภาพหรือคุณภาพไม่ดีจะทำให้เกิดความสิ้นเปลืองและสูญเสียมากกว่าเดิม เกิดผลกระทบต่อการรักษาพยาบาลได้ เป็นที่ทราบกันดีว่าเวชภัณฑ์ชนิดเดียวกันแต่ต่างรุ่นกันก็อาจมีคุณภาพที่ต่างกันได้ และถึงแม้ว่าจะผลิตจากบริษัทเดียวกันแต่ต่างรุ่นกันก็อาจมีคุณภาพต่างกันได้เช่นกัน ดังนั้น การพยายามดำเนินการคัดเลือกให้ได้ว่าเวชภัณฑ์ใดของบริษัทใดมีคุณภาพดีและเหมาะสมจึงเป็นเรื่องที่ต้องให้ความสำคัญ การกำหนดรายละเอียดคุณลักษณะเฉพาะของเวชภัณฑ์จัดได้ว่า เป็นขั้นตอนหนึ่งในการประกันคุณภาพสำหรับการคัดเลือกคุณภาพของผลิตภัณฑ์ที่ต้องการซื้อนั้นด้วย นอกเหนือไปจากการกำหนดเกณฑ์ในการคัดเลือกโรงงานผลิต คุณภาพของผลิตภัณฑ์และการควบคุมคุณภาพหลังการจัดซื้อ
แม้ว่าการดำเนินงานตามมาตรการพัฒนาประสิทธิภาพระบบบริหารเวชภัณฑ์จะได้ดำเนินการมาเป็นระยะเวลาหนึ่งแล้ว แต่ยังคงพบปัญหาในด้านต่าง ๆ อยู่บ้าง ปัญหาหนึ่งของการคัดเลือกคุณภาพเวชภัณฑ์คือ การกำหนดคุณลักษณะเฉพาะของเวชภัณฑ์แต่ละรายการที่จะจัดซื้อซึ่งมีอยู่เป็นจำนวนมาก เนื่องจากการขาดแคลนแหล่งข้อมูลทางด้านวิชาการ ประสบการณ์และความเชี่ยวชาญของเภสัชกรในบางจังหวัด กองโรงพยาบาลภูมิภาคจึงเห็นความจำเป็นในการจัดทำคุณลักษณะเฉพาะของยาของกระทรวงสาธารณสุขเพื่อเป็นบรรทัดฐานให้กับจังหวัดต่าง ๆ ในการนำไปประยุกต์ใช้ได้อย่างสะดวก รวดเร็วและมีประสิทธิภาพมากยิ่งขึ้นต่อไป
2. วัตถุประสงค์
1. เพื่อพัฒนาคุณภาพของการคัดเลือกเวชภัณฑ์ ตามมาตรการพัฒนาประสิทธิภาพระบบบริหารเวชภัณฑ์
ของกระทรวงสาธารณสุข
2. เพื่อจัดทำคุณลักษณะเฉพาะของยาของสำนักงานปลัดกระทรวงสาธารณสุข ซึ่งจะเป็นบรรทัดฐาน
ในการดำเนินงานของสถานบริการต่าง ๆ ในส่วนภูมิภาค
3. ขั้นตอนการดำเนินการ
1. คัดเลือกรายการยาที่จะนำมาจัดทำคุณลักษณะเฉพาะ โดยการรวบรวมข้อมูลจากรายงานการจัดหา
เวชภัณฑ์ร่วมกันในระดับจังหวัดประจำปีงบประมาณ 2541 เพื่อเป็นฐานในการคัดเลือกรายการ
ซึ่งผลการศึกษาข้อมูลดังกล่าว ได้คัดเลือกยาจำนวน 140 รายการแรกที่มีการจัดซื้อร่วมกันมากทั้งประเทศ
มาจัดทำคุณลักษณะเฉพาะ
2. แต่งตั้งคณะทำงานพัฒนาระบบบริหารเวชภัณฑ์ ตามคำสั่งสธ.ที่ 1787 /2542 ลงวันที่ 16 กค. 2542
3. ประชุมคณะกรรมการจัดทำคุณลักษณะเฉพาะของยาเพื่อกำหนดหลักเกณฑ์และรูปแบบในการจัดทำ
คุณลักษณะเฉพาะของยาให้มีความถูกต้องตามหลักวิชาและครอบคลุมประเด็นที่สำคัญ
4. จัดทำคุณลักษณะเฉพาะ(ฉบับร่าง)ของรายการยาที่ได้คัดเลือก เพื่อเป็นแนวทางในการพิจารณา
และปรับปรุงร่วมกัน
5. ประชุมคณะกรรมการจัดทำคุณลักษณะเฉพาะของยาของสำนักงานปลัดกระทรวงสาธารณสุขจำนวน
4 ครั้ง เพื่อแก้ไขและรับรองคุณลักษณะเฉพาะที่จัดทำขึ้น
6. สรุปและเผยแพร่คุณลักษณะเฉพาะของยาแก่หน่วยงานต่าง ๆ ในรูปแบบของแผ่นดิสเก็ตต์และแผ่น
ซีดี ตลอดจนเผยแพร่ผ่านทางเว็บไซต์ของศูนยข้อมูลข่าวสารด้านยา ของกระทรวงสาธารณสุข
7. ติดตาม ประเมินปัญหาจากการจัดทำคุณลักษณะเฉพาะที่กำหนดไว้และปรับปรุงให้เหมาะสมยิ่งขึ้น
4. ระยะเวลาดำเนินการ
ตั้งแต่เดือน กรกฎาคม 2542 - กันยายน 2543
5. ผลการดำเนินงาน
5.1 จัดทำหลักเกณฑ์ในการกำหนดคุณลักษณะเฉพาะของผลิตภัณฑ์ยา เพื่อให้หน่วยงานต่าง ๆ
นำไปปรับปรุงขบวนการจัดซื้อจัดหาเวชภัณฑ์ให้มีประสิทธิภาพ สามารถรองรับภาวะวิกฤตทางเศรษฐกิจที่เกิดขึ้น
โดยเน้นรายการยาที่มีการจัดหาเวชภัณฑ์ร่วมกันในระดับจังหวัด
ในการจัดหาให้ได้มาซึ่งเวชภัณฑ์ที่มีคุณภาพ ราคาเหมาะสมยุติธรรมนั้น จำเป็นต้องอาศัยข้อมูล
คุณลักษณะเฉพาะของผลิตภัณฑ์ (Finished Product Specification) มาประกอบการพิจารณาคัดเลือก เนื่องจาก
ผลิตภัณฑ์แต่ละชนิดอาจมีการกำหนดคุณลักษณะเฉพาะที่แตกต่างกันในแต่ละทะเบียนตำรับที่ขึ้นไว้กับ
สำนักงานคณะกรรมการอาหารและยา ทำให้ผู้ปฏิบัติต้องใช้วิจารณญาณในการตัดสินและแยกแยะความแตกต่างด้านคุณภาพได้อย่างชัดเจน ดังนั้น การจัดทำข้อกำหนดเกี่ยวกับคุณลักษณะเฉพาะของผลิตภัณฑ์ยาขึ้นเพื่อใช้เป็นแนวทางในการคัดเลือกผลิตภัณฑ์จึงเป็นงานที่ส่งผลกระทบต่อขบวนการจัดหาเวชภัณฑ์ที่มีคุณภาพให้กับหน่วยงาน ทั่วประเทศเป็นอย่างมาก เนื่องจากผู้ซื้อสามารถคัดเลือกยาที่มีคุณภาพดี ภายใต้ราคาที่เหมาะสม และที่สำคัญคือเป็นการส่งเสริมอุตสาหกรรมการผลิตยาให้มีการปรับปรุงขบวนการผลิตและแข่งขันด้านคุณภาพมากกว่าการแข่งขันทางด้านราคาเหมือนที่ผ่านมาในอดีต
สำหรับเกณฑ์ที่ใช้ในการกำหนดคุณลักษณะเฉพาะประกอบด้วยสาระสำคัญ ดังนี้
5.1.1 มาตรฐานอ้างอิง ได้เลือกใช้ตำรายาที่กระทรวงสาธารณสุขประกาศ (ฉบับล่าสุด) เป็นมาตรฐานอ้างอิงในการกำหนดคุณลักษณะเฉพาะของยาเป็นหลัก เนื่องจากตำรายาแต่ละฉบับที่ออกมาใหม่จะมีการพัฒนา
ข้อกำหนดของผลิตภัณฑ์ให้ทันสมัยอย่างต่อเนื่อง ประกอบกับทะเบียนยาของประเทศไทยเป็นทะเบียนตลอดชีพ (เว้นแต่จะมีการประกาศตามกฎหมายให้เพิ่มเติมสาระสำคัญ หรือยกเลิกทะเบียนเป็นรายกรณี) ดังนั้น ข้อกำหนดในทะเบียนที่ได้รับอนุญาตมานานแล้ว อาจมีข้อกำหนดบางรายการที่ตำรายาฉบับใหม่ได้มีการปรับปรุงแก้ไขให้รัดกุมมากขึ้น แต่ทะเบียนของผู้ผลิตยังมิได้ดำเนินการแก้ไข ดังนั้น การใช้มาตรฐานอ้างอิงจากตำรายาที่กระทรวงสาธารณสุขประกาศ (ฉบับล่าสุด) จึงเท่ากับเป็นการกระตุ้นให้ผู้ผลิตมีความตระหนักและระมัดระวังในเรื่องคุณภาพของผลิตภัณฑ์มากขึ้นและเกิดการพัฒนาทะเบียนตำรับให้มีความทันสมัยและถูกต้องตามหลักวิชาอยู่เสมอ
ตำรายาที่กระทรวงสาธารณสุขประกาศรับรอง (ฉบับล่าสุด) คือประกาศเรื่องระบุตำรายา ฉบับที่ 4 พ.ศ. 2539 และฉบับที่ 5 พ.ศ. 2541 ซึ่งประกอบด้วยตำรายา ดังนี้
1. Thai Pharmacopoeia Volume I Part 1,Part 2
2. International Pharmacopoeia 3rd
3. The United States Pharmacopoeia 22nd revision, The National Formulary, 17th
4. The United States Pharmacopoeia 23nd revision, The National Formulary, 18th
5. British pharmacopoeia 1998
6. British pharmacopoeia 1993
7. British pharmacopoeia (Veterinary ) 1985
8. Thai Pharmacopoeia Vol. II Part 1
9. Thai Herbal Pharmacopoeia Vol. I
สำหรับตำรายาที่นิยมใช้ในการอ้างอิงสำหรับการกำหนดเกณฑ์มาตรฐานของผลิตภัณฑ์ยาสำเร็จรูปส่วนใหญ่ คือ USP และ BP (& European Pharmacopoeia) ซึ่งหัวข้อในตำรายาแต่ละฉบับจะมีความแตกต่างกันในรายละเอียด ตัวอย่างเช่น การกำหนดเกณฑ์มาตรฐาน Content Uniformity ของ USP และ BP พบว่าการกำหนดเกณฑ์สูงสุด และต่ำสุดในแต่ละหน่วยเปรียบเทียบแตกต่างกัน โดยใน USP จะกำหนดจากปริมาณที่แจ้ง ส่วน BP จะกำหนดจากปริมาณที่ตรวจพบ นอกจากนี้ ใน USP.ยังกำหนด %RSD (Relative Standard deviation) ไว้ด้วย (รูปที่ 1,2)
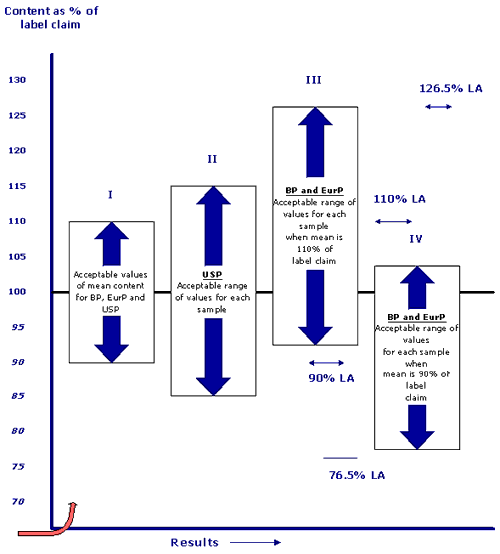

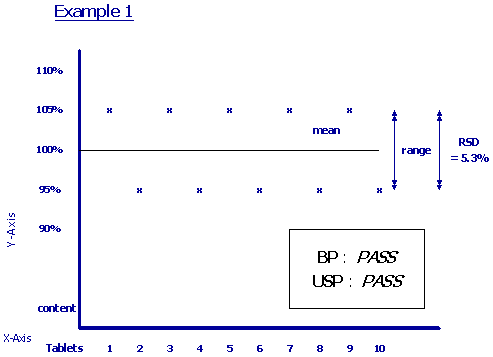
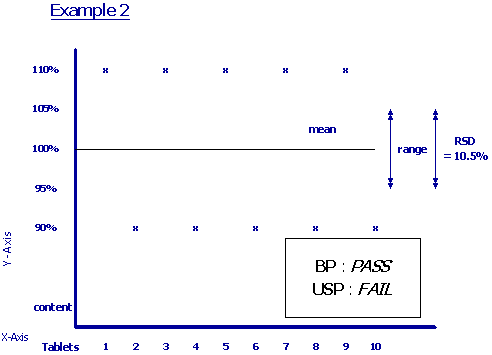
ในการพิจารณาคุณภาพยาหรือเภสัชภัณฑ์ตามเกณฑ์มาตรฐานจากเอกสารแสดงผลการตรวจวิเคราะห์ (Certificate of Analysis) เพื่อเปรียบเทียบคุณภาพของผู้ผลิตแต่ละรายการเป็นสิ่งที่กระทำได้ยาก จำเป็นต้องอาศัยพื้นฐานความเข้าใจในแต่ละข้อว่าถูกกำหนดขึ้นเพื่อวัตถุประสงค์ใด ซึ่งความเข้าใจดังกล่าวอาจเป็นเพียงแนวทางขั้นต้นที่ช่วยในการพิจารณาคัดเลือกผลิตภัณฑ์ ส่วนเรื่องความน่าเชื่อถือของเอกสารแสดงผลการตรวจวิเคราะห์ (Certificate of Analysis) นั้น นอกจากการยืนยันโดยการตรวจวิเคราะห์ทางห้องปฏิบัติการแล้ว ยังต้องมีการสุ่มตรวจ GMP (Good Manufacturing Practice) ในโรงงานเพื่อพิสูจน์ว่าผู้ผลิตได้มีการตรวจสอบคุณภาพยาที่ผลิตในแต่ละรุ่นจริงตามที่ได้รับ GMP หรือไม่ มิใช่เพียงแต่ดูใบรับรอง GMP เท่านั้น
5.1.2 วิธีการกำหนดคุณเฉพาะของยา ในกรณีที่ตัวยาและรูปแบบยานั้นมีระบุไว้ในตำรายา จะถือว่า ข้อกำหนดตามตำรายาเป็น Minimum Requirements และหากว่ามีการกำหนดคุณลักษณะเฉพาะไว้ในตำรายาหลายเล่ม และมีหัวข้อไม่เท่ากัน จะใช้ข้อมูลทางวิชาการในการปรับให้สอดคล้องกัน ทั้งนี้ จะไม่มีการกำหนดหัวข้อใด ๆ
เพิ่มเติม เกินกว่าที่ตำรายาเหล่านั้นระบุไว้
ในกรณีที่ตัวยาหรือรูปแบบยานั้นไม่มีระบุไว้ในตำรายาใดๆ หรือมีระบุเฉพาะวัตถุดิบของตัวยานั้น จะใช้ข้อกำหนดตาม General Requirements ของรูปแบบยานั้นตามที่ตำรายากำหนดไว้เป็นมาตรฐาน
5.1.3 หัวข้อหลักในการควบคุมคุณภาพ แต่ละผลิตภัณฑ์จะประกอบด้วยเนื้อหาหลัก 4 ส่วน คือ
1) ชื่อยา จะกำหนดชื่อยา (Generic Name) พร้อมรูปแบบของยา ( Dosage Form) ตรงตามที่ระบุไว้ในตำรายา
2) คุณสมบัติทั่วไป เป็นลักษณะ (Appearance) ของผลิตภัณฑ์ และกำหนดรายละเอียดอื่นๆ ตามลำดับ ดังนี้
- รูปแบบ : ยาเม็ด,ยาน้ำ,ยาฉีด,ยาพ่น เป็นต้น
- ส่วนประกอบ : ตัวยาสำคัญที่เป็นส่วนผสมของผลิตภัณฑ์ โดยระบุปริมาณตัวยาสำคัญและรูปแบบเกลืออย่างชัดเจน (ถ้ามี)
- ภาชนะบรรจุ : ลักษณะของบรรจุภัณฑ์ตามที่ระบุไว้ในตำรายาหรือเอกสารอ้างอิงทางวิชาการอันมีผลต่อคุณภาพหรือความคงตัวของยาอย่างมีนัยสำคัญ
- ฉลาก : มี Labelling ถูกต้องตามมาตรฐานที่กำหนดไว้ในตำรายา และเงื่อนไขอันเกี่ยวเนื่องกับอายุการใช้งานของผลิตภัณฑ์ เช่น วันหมดอายุ ( Expired Date), วันผลิต (Manufacturing Date) เป็นต้น
3) คุณสมบัติทางเทคนิค เป็นหัวข้อในการควบคุมคุณภาพของผลิตภัณฑ์นั้นๆตามเกณฑ์
มาตรฐาน ซึ่งในที่นี้หมายถึง มาตรฐานของรูปแบบยาเตรียมที่มีระบุในตำรายาหรือมาตรฐานที่ผู้ผลิตกำหนดขึ้นตาม general requirements
General requirements ของ finished product แต่ละรูปแบบ นอกจากการตรวจเอกลักษณ์ และหาปริมาณแล้ว การขึ้นทะเบียนตำรับยาจะกำหนดหัวข้ออื่นๆ ดังตัวอย่างต่อไปนี้
| USP 23 | ||
|
Tablet, capsule
|
|
Injection
|
|
|
|
|
Cream, Ointment, Gel, Lotion
|
|
Sterile powder
|
|
|
|
|
Syrup, Suspension (oral solution)
|
|
ยาตาเช่น Eye lotion
|
|
|
|
|
Cream, Ointment, Gel, Lotion
|
|
Sterile powder
|
|
|
|
|
Inhaler
|
|
Aerosol
|
|
|
|
| BP 1999 | ||
|
Eye Preparation
|
|
Parenteral Preparation
|
|
|
|
|
Inhalation
|
|
Rectal Preparation
(suppo., rectal solution, enema etc.) |
|
|
|
|
Oral liquids (solution, elixir, mixture, oral drops)
|
|
Extracts (liquid extracts)
|
|
|
|
|
Spirit
|
|
Soft Extract
|
|
|
|
|
Transdermal Patches
|
|
Tincture
|
|
|
|
|
Vaginal Preparation
|
|
Dry Extract
|
|
|
|
|
Urethral Stick
|
|
|
|
|
|
5.1.4 เงื่อนไขอื่นๆ ซึ่งเกี่ยวข้องกับเอกสารประกอบในการเสนอราคาเพื่อจัดซื้อผลิตภัณฑ์เช่น เอกสารเกี่ยวกับการขึ้นทะเบียน เอกสารเกี่ยวกับการรับรองมาตรฐานบริษัทผู้ผลิต นอกจากนั้น ในผลิตภัณฑ์บางรายการ ควรเพิ่มการพิจารณาใบตรวจวิเคราะห์(Certificate of Analysis ) ของบริษัทผู้ผลิตวัตถุดิบ เพื่อประกอบการตัดสินใจควบคู่กับการดูผลตรวจวิเคราะห์ของผลิตภัณฑ์สำเร็จ (Finished Product ) เนื่องจากคุณลักษณะเฉพาะบางอย่างมีกำหนดให้ตรวจวิเคราะห์เฉพาะในวัตถุดิบและจากหลักการที่ว่าผลิตภัณฑ์ที่ดีควรมาจากกระบวนการผลิตที่ดี (Good Manufacturing Practice) ซึ่งต้องมีการวิเคราะห์วัตถุดิบทุกครั้งก่อนการผลิตยา
ตัวอย่างของผลิตภัณฑ์ที่มีการเพิ่มเติมเงื่อนไขอื่นๆ เช่น กรณีของ Blood Products ที่กำหนดว่า ต้องตรวจไม่พบเชื้อHepatitis B, Hepatitis C ,HIV ในวัตถุดิบที่ใช้ในการผลิต หรือกรณีการตรวจหา Related Substances ในยาบางชนิด ซึ่งตำรายาระบุให้ตรวจวิเคราะห์ตามมาตรฐานที่กำหนดของวัตถุดิบที่จะใช้ในการผลิตยา หรือกรณีของยากลุ่มต้านจุลชีพซึ่งผลการตรวจวิเคราะห์ Potency ของวัตถุดิบ จะช่วยให้ผู้ซื้อสามารถแยกแยะ
คุณภาพของวัตถุดิบที่ผู้ผลิตเลือกใช้ในการผลิตได้ ซึ่งในผลิตภัณฑ์สำเร็จรูปเหล่านี้ อาจไม่มีข้อกำหนดให้ผู้ซื้อสามารถพิจารณาได้เพราะมีกำหนดไว้เฉพาะในวัตถุดิบ
6. ความสำคัญของข้อกำหนดหรือเกณฑ์มาตรฐาน
ในการจัดซื้อให้ได้มาซึ่งผลิตภัณฑ์ยาที่มีคุณภาพในราคาที่เหมาะสม จำเป็นอย่างยิ่งที่ผู้จัดซื้อจะต้องเข้าใจถึงวัตถุประสงค์ในการกำหนดเกณฑ์มาตรฐานแต่ละหัวข้อ จึงจะสามารถกำหนดคุณลักษณะเฉพาะของผลิตภัณฑ์ได้อย่างถูกต้องและเหมาะสม มีความครอบคลุมประเด็นสำคัญ ในการกำหนดคุณภาพของผลิตภัณฑ์นั้นๆ
6.1 ข้อกำหนดที่ควรทราบเกี่ยวกับยาสำเร็จรูป
6.1.1 Identification เป็นการยืนยันเอกลักษณ์ของสารว่าตรงตามที่แจ้งไว้บนฉลาก ในบางครั้ง purity test ก็สามารถใช้ในการตรวจเอกลักษณ์ด้วย เช่น การตรวจ Water content ของ สารที่อยู่ในรูปของ hydrates, การตรวจ optical rotation ของ optical isomer.
6.1.2 Label amount
เป็นการตรวจหาปริมาณตัวยาสำคัญในผลิตภัณฑ์สำเร็จรูปว่ามีตัวยาสำคัญอยู่ในช่วงที่แจ้งไว้ตามทะเบียนหรือไม่ หัวข้อดังกล่าวเป็นหัวข้อที่สำคัญมากเนื่องจากปริมาณตัวยาสำคัญเป็นส่วนหนึ่งที่จะบ่งบอกถึงคุณภาพของผลิตภัณฑ์ว่ายังสามารถนำไปใช้ในการรักษาได้หรือไม่
ค่าที่ใช้ในการบอกปริมาณตัวยาสำคัญ มักระบุเป็นเปอร์เซ็นต์และจะต้องพิจารณาเกี่ยวกับการกำหนดรายละเอียดของตัวยาสำคัญในผลิตภัณฑ์ว่าอยู่ในรูปเบสหรือในรูปของเกลือ ในกรณีที่ตัวยาสำคัญอยู่ในรูปของเกลือ จะต้องระบุถึงค่าสมมูลกับตัวยาที่อยู่ในรูปเบสบนฉลาก ยกเว้นผลิตภัณฑ์บางรายการที่กำหนดเปอร์เซ็นต์ปริมาณตัวยาสำคัญในรูปของเกลือด้วย ตัวอย่างเช่น Dopamine HCl
6.1.3 น้ำหนักเฉลี่ยต่อหน่วย (weigh/unit)
โดยปกติ ผู้ผลิตได้ขึ้นทะเบียนตำรับยาไว้โดยระบุสูตรตำรับและน้ำหนักต่อหน่วยตามสูตร พร้อม range of mean weight บ่อยครั้งที่พบว่าตัวอย่างที่ตรวจวิเคราะห์มีน้ำหนักต่อหน่วยไม่อยู่ในช่วงที่ระบุไว้ในทะเบียนยา (ทย.1) หรือลักษณะผงยา,แกรนนูล, ไม่ตรงตามที่ระบุไว้
6.1.4 Uniformity of dosage units เป็นการตรวจสอบหาความสม่ำเสมอของตัวยาในแต่ละหน่วย ซึ่งโดยทั่วไป มีเกณฑ์การกำหนดไว้ 2 ลักษณะ คือ
1) ความแตกต่างจากน้ำหนักเฉลี่ย (Uniformity of weight หรือ Weight variation) โดยส่วนใหญ่จะเลือกใช้ uniformity of weight ในกรณีที่ยามีปริมาณตัวยามากและเลือกใช้ content uniformity ในกรณี
ที่มีตัวยาน้อย
การหาความแตกต่างจากน้ำหนักเฉลี่ย เป็นการนำแต่ละหน่วยของผลิตภัณฑ์ยามาชั่งน้ำหนักเพื่อหาค่าน้ำหนักเฉลี่ยและน้ำหนักแต่ละหน่วย โดย USP และ BP มีมาตรฐานในการกำหนดค่าเบี่ยงเบนของแต่ละหน่วยแตกต่างกันดังนี้
| USP 23 | ||||
|
น้ำหนักตัวยา
|
|
Weight variation
|
||
|
> 50 mg |
|
|
||
| BP 1999 | ||
|
น้ำหนักตัวยา
|
|
Weight variation
|
|
80 mg หรือน้อยกว่า |
|
 5 % 7.5 % 10 % 5 % 7.5 % 10 % |
ข้อควรระวัง การตรวจสอบ Weight variation หรือ uniformity of weight จะใช้ในผลิตภัณฑ์สำเร็จรูปชนิดแข็ง (Solid dosage form) กรณีที่เป็นยาเม็ดจะกำหนดในยาเม็ดชนิด Plain tablet หรือ filmed coated tablet แต่ในกรณีของ sugar coated tablet ไม่กำหนดเป็น Uniformity of weight หรือ Weight variation แต่กำหนด Content uniformity แทน
2) ความสม่ำเสมอของตัวยาแต่ละหน่วย (Content uniformity)
การกำหนดให้ทดสอบหาความสม่ำเสมอของตัวยาแต่ละหน่วย (Content uniformity) เริ่มมีกำหนดครั้งแรกใน USP XVII ในปัจจุบัน การกำหนดให้ต้องหาความสม่ำเสมอของตัวยาแต่ละหน่วย ตำรายาแต่ละฉบับมีหลักเกณฑ์พิจารณาโดยดูจากปริมาณของตัวยาสำคัญซึ่งอยู่ในยาแต่ละหน่วย ดังนี้
USP - กำหนดให้หา Content uniformity ในตำรับที่มีตัวยาสำคัญเท่ากับหรือน้อยกว่า 50 มิลลิกรัม
Ph. Eur - กำหนดให้หา Content uniformity ในตำรับที่มีตัวยาสำคัญเท่ากับหรือ น้อยกว่า 2 มิลลิกรัม
หรือ 2%w/w
TP - กำหนดให้หา Content uniformityเหมือนกับ BP. , Ph. Eur
6.1.5 เวลาในการกระจายตัวของยา (Disintegration time)
เป็นการหาการกระจายตัวของยาสำเร็จรูป เพื่อควบคุมการผลิตยาในแต่ละBatch ว่ายังมีความสม่ำเสมอในเกณฑ์ที่กำหนดไว้หรือไม่ การหา Disintegration time เป็นการทดสอบว่ายาสำเร็จรูปนั้นกระจายตัวในสภาวะที่กำหนด และปลดปล่อยตัวยาออกมาในเวลาที่กำหนดไว้หรือไม่ เป็นการทดสอบยาสำเร็จรูปที่ให้ทางปาก แต่กรณีที่เป็นยาเม็ดหรือแคปซูลที่ใช้อมหรือเคี้ยวก่อนกลืน หรือ Modified-release ไม่ต้องตรวจหา Disintegration
สำหรับ Modified-release tablet หรือ capsule เช่น enteric-coated tablet จะทำ Drug release แทน โดยใช้ medium เป็น 0.1 M HCL (เลียนแบบสภาวะในกระเพาะอาหาร) และ Buffer pH 6.8 (เลียนแบบสภาวะในลำไส้)
เนื่องจากในการดูดซึมของยานั้น จะต้องผ่านการละลายในรูปสารละลายก่อน และก่อนการละลายต้องมีการกระจายตัวเป็น Particles ซึ่งเวลาที่ใช้ในการกระจายตัวหมดจะกำหนดไว้ในยาแต่ละแบบ สิ่งที่ควรคำนึงคือ เวลาในการกระจายตัวของยา (in vitro) ไม่จำเป็นว่าจะต้องสัมพันธ์กับ in vivo action ของ solid dosage form นั้นๆ
ในการศึกษาเปรียบเทียบเวลาในการกระจายตัวของยากับอัตราเร็วในการละลายของตัวยา (dissolution rate) หรืออัตราเร็วของ Initial absorption ของยาเม็ดแอสไพรินหลายๆ ผู้ผลิต (จากเอกสาร WHO) พบว่า ยาเม็ดที่ใช้เวลาในการกระจายตัวยาวนานกว่าตำรับอื่นกลับมีการดูดซึมเร็วกว่า (the faster absorbed tablets had the longer disintegration time) ดังนั้นการใช้เวลา (นาที) ของการกระจายตัวของยา มาเปรียบเทียบคุณภาพของยาแต่ละผู้ผลิต ควรคำนึงถึงเหตุผลดังกล่าวด้วย และการรายงานผลในหัวข้อนี้ว่า เข้า หรือ ผิดมาตรฐานตามเกณฑ์กำหนด น่าจะเหมาะสมกว่าการระบุเป็นตัวเลข เพื่อไม่ให้เกิดการเข้าใจผิดและนำไปเปรียบเทียบคุณภาพทาง in vitro ยกเว้นจะมีผลการศึกษาทาง in vivo มาสนับสนุน
6.1.6 การละลายของตัวยา ( Dissolution)
เป็นการทดสอบการละลายของตัวยาเพื่อจำแนกคุณภาพของผลิตภัณฑ์และปัจจัยในการตั้งสูตรตำรับ ดังนั้น การทดสอบการละลายของตัวยาจึงเป็นหัวข้อหนึ่งที่ใช้ประกอบในการวิจัยและพัฒนาผลิตภัณฑ์ยา ไม่ว่าเป็นการศึกษาเปรียบเทียบระหว่างสูตรตำรับที่กำลังพัฒนา หรือเปรียบเทียบกับ reference products ก็ตามและสภาวะการทดสอบ dissolution ที่ดีควร แยกความแตกต่างในการละลายในเวลาต่างๆ กัน ของแต่ละสูตรหรือแต่ละ product ได้ ตัวอย่างเช่น Ibuprofen Tablet ใน USP 23 กำหนดสภาวะการทดสอบไว้แบบหนึ่งต่อมา USP ได้มีการแก้ไขสภาวะการทดสอบใน USP 23 ฉบับแก้ไขเพิ่มเติม โดยใช้วิธีของ US.FDA
|
USP 23 (เดิม)
|
USP 23.5 th supplement
|
|
| Medium : | Phosphate | buffer pH 7.2 same |
| Apparatus : | basket | paddle |
| ความเร็วรอบ/นาที | 150 | 50 |
| ใช้เวลาในการทดสอบ : | 30 นาที | 60 นาที |
| Tolerance (Q) : | 70% la | 80% la |
สภาวะการทดสอบทั้งสองแบบพบว่าที่เวลาต่างๆกัน ในยาเม็ดบริษัทเดียวกัน สภาวะการทดสอบของ USP 23 (เดิม) จะไม่เห็นความแตกต่างของปริมาณตัวยาที่ละลายออกมาได้ชัดเจน เหมือนวิธีใหม่ ดังนั้น
จึงมีการเปลี่ยนแปลงสภาวะการทดสอบใน USP 23 ไปตามวิธีของ US.FDA
เมื่อมีการเปลี่ยนแปลงดังกล่าว ผู้ผลิตควรเปลี่ยนแปลงมาตรฐานในทะเบียนยาให้เป็นปัจจุบันตามตำรับยาที่มีการแก้ไขและผู้จัดซื้อเองก็ควรกำหนดคุณลักษณะเฉพาะของ Ibuprofen Tablet ในหัวข้อ Dissolution ให้เป็นไปตาม USP 23 Supplement ทั้งนี้ เพื่อเป็นแนวทางในการคัดเลือกผลิตภัณฑ์ที่มีความมั่นใจในคุณภาพมากขึ้นและเป็นการกระตุ้นบริษัทผู้ผลิตให้มีความระมัดระวังเกี่ยวกับคุณภาพของผลิตภัณฑ์ให้มีความถูกต้องตามหลักวิชาการอยู่เสมอ
6.1.7 Related Substances
Related Substances คือ สารสลายตัวหรือสาร intermediates และ by-product ที่ได้จากการสังเคราะห์สารอินทรีย์ รวมถึงสาร co-extracted substances จากสารธรรมชาติสมุนไพร รวมทั้ง degradation products ของสารหรือตัวยา
ส่วน residual organics, solvents, น้ำ และ inorganic anions/cations สิ่งตกค้างจาก cell, จุลินทรีย์และอาหารเลี้ยงเชื้อในการหมัก ไม่จัดเป็น Related Substances
การควบคุมเกี่ยวกับ Related Substances ทำให้เราสามารถทราบ impurity ของ synthetics route และทราบ likely decomposition pathways
ในการศึกษาความคงสภาพทางด้านเคมีของผลิตภัณฑ์ยา เพื่อกำหนดอายุของผลิตภัณฑ์นั้น ๆ วิธีตรวจวิเคราะห์ในการหาปริมาณตัวยาสำคัญจะต้องแสดงให้เห็นได้ชัดเจนว่า วิธีที่ใช้สามารถแยกตัวยาสำคัญออกจาก Related Substances ได้ รวมทั้งสารอื่นๆในสูตรตำรับ ถ้าในการหาปริมาณตัวยาสำคัญที่ไม่มีหัวข้อการตรวจหาสารสลายตัว แสดงว่าวิธีการตรวจวิเคราะห์นั้นไม่สามารถใช้ในการศึกษาความคงสภาพของยาตำรับนั้น ๆ ดังนั้น การกำหนดวันหมดอายุบนฉลากได้ถูกต้อง จำเป็นต้องทราบเกี่ยวกับ Related Substances ของตัวยานั้นๆด้วย
การตรวจวิเคราะห์เกี่ยวกับ Related Substances อาจระบุเป็นปริมาณ (%) ในกรณีที่เป็นวิธีวิเคราะห์ที่หาปริมาณได้ เช่น HPLC แต่ถ้าเป็นวิธีวิเคราะห์ที่เป็นการเปรียบเทียบความเข้มของสี เช่น TLC หรือ Colorimetry ผลอาจระบุได้เพียง เข้า หรือ ผิดมาตรฐานเท่านั้น สำหรับใน USP ส่วนใหญ่จะกำหนดหัวข้อการหา
สารสลายตัวไว้ในการตรวจวิเคราะห์วัตถุดิบและหลายรายการที่ไม่มีการกำหนดไว้ใน finished product ทั้งนี้ อาจเนื่องจากวิธีวิเคราะห์หาปริมาณใน finished product สามารถแยกสารอื่นที่ไม่ใช่ตัวยาสำคัญออกไปได้ ซึ่งแตกต่างกับการกำหนดมาตรฐานใน BP ที่วิธีหาปริมาณตัวยาสำคัญ ยังนิยมใช้วิธีพื